CO2 electroreduction (CO2ER) powered by clean electricity provides a promising route to close the anthropogenic carbon cycle and store intermittent renewable energy in chemicals or fuels. Relative to the conventional CO2ER system that relies on alkaline/neutral electrolytes, acidic CO2ER offers enhanced carbon utilization efficiency (>50%) and energy efficiency, attributed to the theoretically inhibited carbonation reactions and the reduced energy penalty for (bi)carbonate regeneration. Unfortunately, the acidic CO2ER generally confronts the strong competition from hydrogen evolution reaction (HER) due to the high proton activity. A solution to suppress HER is to introduce alkali metal cations (e.g., K+ and Cs+) into the acidic electrolyte. These alkali metal cations can decrease the local availability of protons and enhance the reaction kinetics toward the carbon-based products by optimizing the nature of electric double layer (EDL). However, such a system often faces the problem of salt precipitation on the gas diffusion electrode (GDE) due to the local alkalinity and concentrated alkali metal cations under operation. The deposited (bi)carbonate salts could gradually obstruct the CO2 diffusion channels of GDE, diminish the GDE hydrophobicity, and induce the electrode flooding, which ultimately results in a poor operational stability. To simultaneously suppress HER and salt precipitation, very recently one alternative solution is to immobilize the cationic groups on the catalyst surface, which prolonged the lifetime up to 150 h.
As we know, organic cations, with large ion size, hydrophobicity and unique solvation shell structures, are another important category of cations in electrochemistry. Relative to metal cations, the tunable structures of organic cations could offer more opportunities to regulate the interfacial microenvironment by virtue of the intermolecular interaction among their functional groups, water, and other reactive/non-reactive species in the EDL. These features make them a highly promising substitute for conventional alkali metal cations in electrolytes for CO2ER. In principle, owing to their cationic nature, organic cations in acidic electrolyte could shield the electric field from the cathode by accumulating on the Helmholtz plane, thereby suppressing the migration of hydronium ions and shifting the selectivity from HER to CO2ER. Nevertheless, to date, no studies have demonstrated that organic cations can solely drive the acidic CO2ER. Furthermore, the molecule-level understanding of how these organic cations affect the CO2ER process lags far behind that of metal cations. This knowledge gap arising from more intricate dynamic behavior of organic cations within the EDL hampers further advancement in designing metal-cations-free systems.
Building on the above analyses, Prof. Hongwen Huang et al. propose that an ideal organic cation should both possess high solubility in the corresponding bicarbonate salt and have the smallest possible size to prevent salt precipitation and minimize HER kinetics. Following this principle, tetramethylammonium cations (TMA+) with a radius of 3.47 Å are chosen as the supporting electrolyte in acid. Meanwhile, the solubility of TMA+-bicarbonate salt can reach ~12.4 M, which is 3.4 times higher than that of KHCO3 (3.62 M at 20 oC). Through this strategic cation engineering, we establish an acidic CO2ER system achieving a high product selectivity nearing 100% and a super-long lifetime exceeding 2,600 h at an industrially relevant current density. The effectiveness of this design principle is even more validated through a 1,200-hour stable alkaline CO2ER operation. Mechanistic investigations reveal that TMA+ cations mainly work by disrupting the hydrogen-bond networks for proton shuttling to suppress HER in acid, meanwhile the hydrophobicity of TMA+ and much higher bicarbonate solubility synergistically prevent the salt precipitation, which is different from the cation effect of conventional alkali metal cations. This work not only introduces an advanced paradigm for propelling the CO2 electrolysis toward practical applications, but also broadens our understanding of the cation effect.
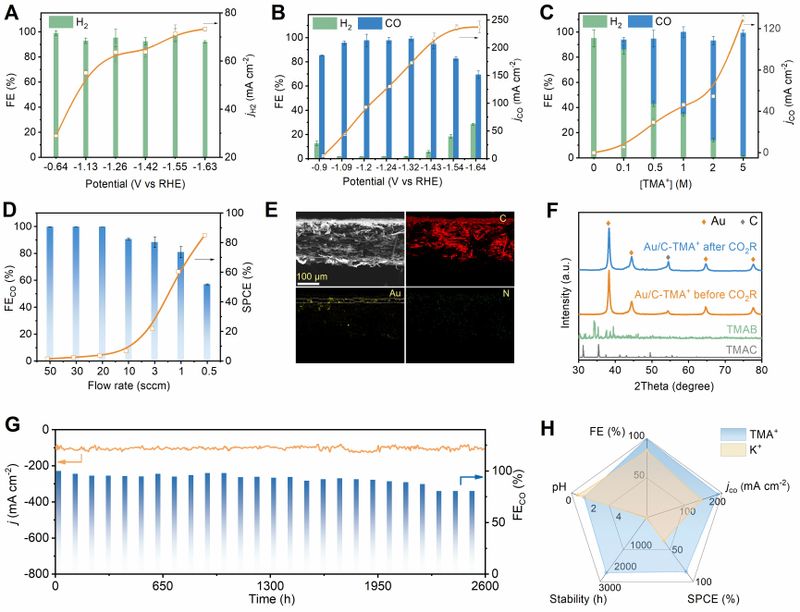
Figure 1. TMA+-mediated acidic CO2ER performance. (A) CO2ER FE (left axis) and H2 current density (right axis, jH2) over Au/C catalyst at different potentials in 0.05 M H2SO4 electrolyte. All FE and j values are means with standard deviations as error bars based on three replicates. (B) FE and CO partial current density (jCO) in 0.05 M H2SO4 electrolyte with 5 M TMA+. (C) FE and jCO in acidic electrolyte containing different concentrations of TMA+ at -1.23 VRHE. (D) CO FE and SPCE at different CO2 flow rates in 0.05 M H2SO4 electrolyte with 5 M TMA+ at 200 mA cm-2. (E) SEM image and the corresponding EDS mapping of the cross section of GDE after CO2ER operation. Scale bar, 100 μm. (F) XRD of GDE before and after CO2ER operation. The XRD patterns of tetramethylammonium chloride (TMAC) and tetramethylammonium bicarbonate (TMAB) are also given for reference. (G) Operation stability at -1.23 VRHE. (H) Comparison of FE, jCO, SPCE, stability lifetime, and electrolyte pH for acidic CO2ER steered by different cations.
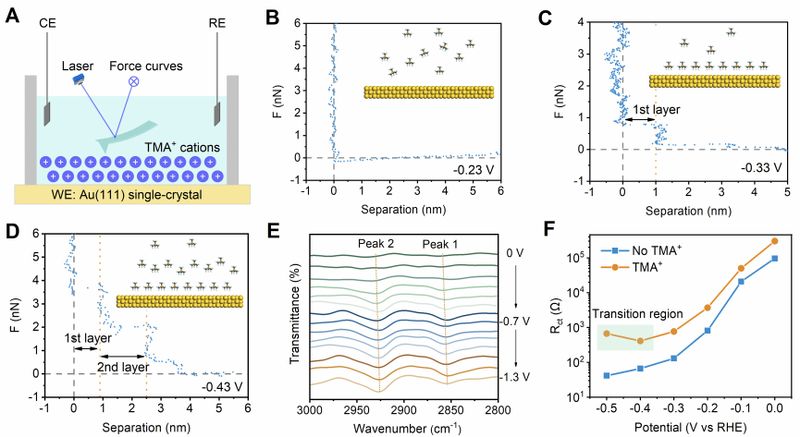
Figure 2. Potential-dependent dynamic assembly of TMA+ in the EDL. (A) Schematic illustration of EC-AFM to probe the interfacial dynamic of TMA+ on Au(111) single-crystal electrode. CE, RE, and WE represent the counter electrode, reference electrode, and working electrode, respectively. (B)-(D) Force-separation curves of TMA+ on Au(111) surface collected at -0.23 VRHE (B), -0.33 VRHE (C) and -0.43 VRHE (D). The electrolyte comprises 0.05 M H2SO4 and 5 M TMA+ aqueous solution. The inserted illustrations depict the corresponding interfacial configuration of TMA+ at different potentials. (E) ATR-FTIRS spectra of Au electrode in 0.05 M H2SO4containing 5 M TMA+. (F) The RCT of Au/C catalyst in acidic electrolyte without or with 5 M TMA+. The data were derived by fitting the EIS plots.
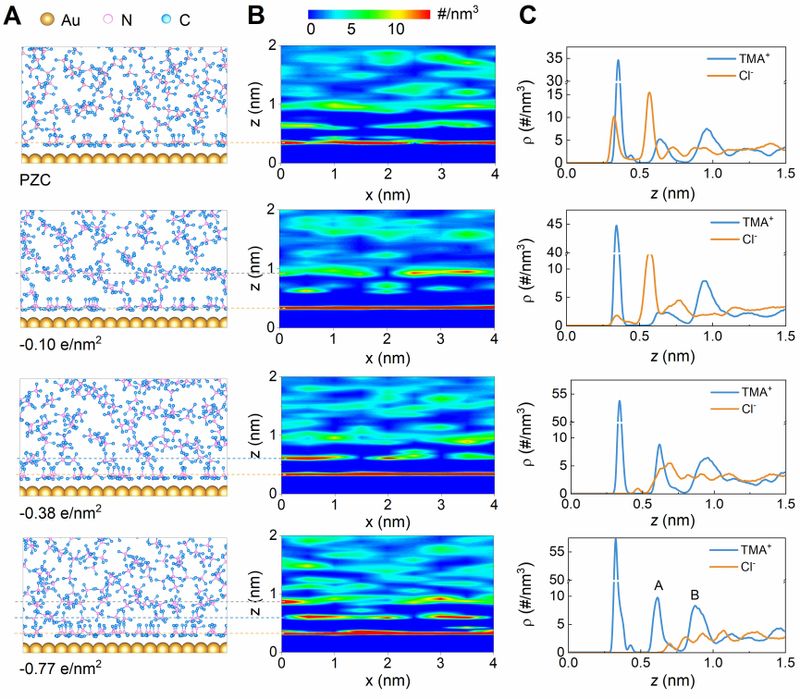
Figure 3.MD simulations on dynamic behavior of TMA+ at varying charge densities. (A) MD snapshots of the system. (B) The corresponding density maps of TMA+ in x-z plane. (C) The corresponding distribution profiles of TMA+ and Cl- ions (from tetramethylammonium chloride electrolyte) along the z-axis.
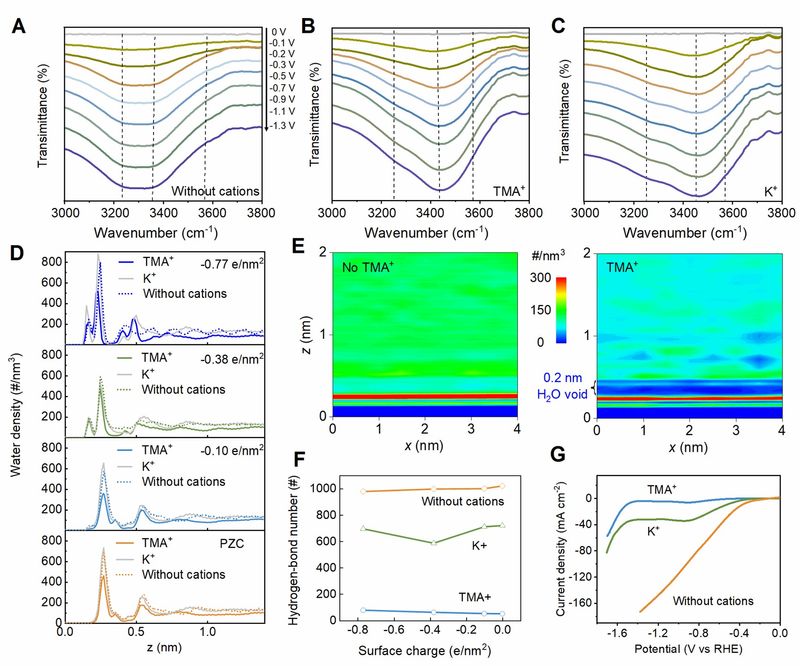
Figure 4.Interfacial microenvironment tailored by TMA+. (A)-(B) In-situ ATR-FTIRS spectra of interfacial water in acidic electrolyte without cations (A), with TMA+ (B), and with K+ (C). (D) Distribution density profiles of interfacial water in acidic electrolyte with different cations along z-direction. (E) Two-dimensional mapping of interfacial water in acidic electrolyte with or without TMA+ at -0.38 e/nm2. (F) Hydrogen-bond number of interfacial water in acidic system with or without cations. (G)HER LSV curves of Au/C catalyst in acidic electrolyte with and without cations. The data are collected on rotating disk electrode in a single cell at a rotating rate of 0 rpm.
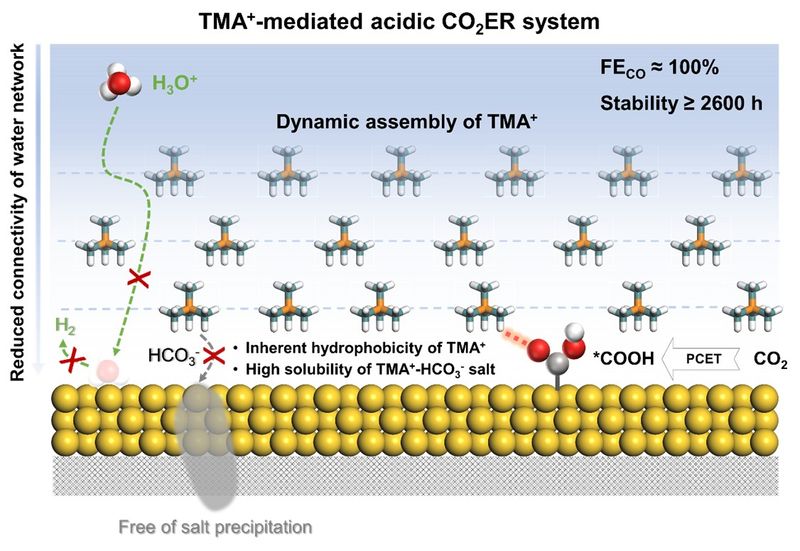
Figure 5. Mechanism illustration of acidic CO2ER system mediated by TMA+ cations. This shows how TMA+ suppresses HER, enhances CO2 reduction kinetics, and prevents salt precipitation.
The related paper entitled “Dynamic assembly of interfacial organic cations enables highly stable and selective CO2 electroreduction in acid” has been published in Science Advances on April 29, 2026 (Paper link: https://www.science.org/doi/10.1126/sciadv.aea1941, DOI: 10.1126/sciadv.aea194). Dr. Wenchuan Lai, Dr. Yan Qiao and Dr. Shuai Liu are the first authors. Prof. Hongwen Huang and Prof. Zheng Hu from our department are co-corresponding authors. This work was jointly supported by the National Natural Science Foundation of China (grants 22322902, U22A20396, 22211540385), and National Key Research and Development Program of China (grant 2021YFA1502000).